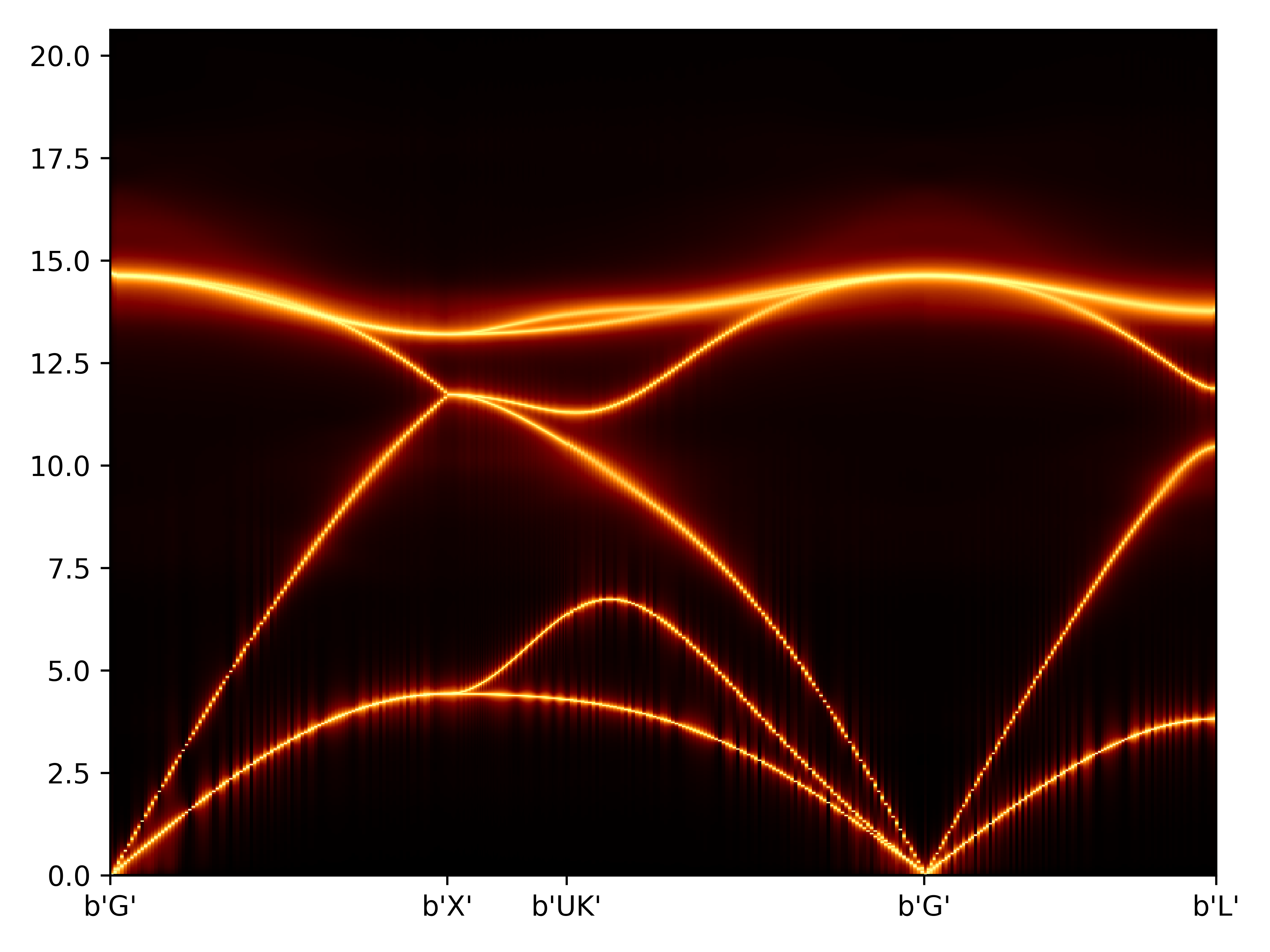
TDEP计算高温声子色散
lammps
输入文件
in.lammps #lammps计算的输入文件
data.cell #lammps计算需要的超胞文件,可以直接在in.lammps中设置
grep_dump.sh #处理lammps输出的dump信息,得到TDEP需要的输入文件
MTP100.mtp #LAMMPS计算需要的势文件,根据需要选取不同种类,不唯一
#TDEP所需要的文件
infile.ucposcar #原胞
infile.meta #TDEP需要的分子动力学的一些信息,结构数目,温度,步长等
infile.ssposcar #TDEP计算需要的初始超胞
infile.positions #TDEP计算需要的每步的原子坐标信息
infile.forces #TDEP计算需要的每步的原子受力
infile.stat #TDEP计算需要的每步的能量 温度 压强 应力等信息
1 LAMMPS计算
# molecular dynamics NVT
units metal
boundary p p p
atom_style atomic
read_data data.pos
pair_style MLIP mlip.ini
pair_coeff * *
variable t equal 1113
neighbor 0.3 bin
timestep 0.001
thermo_style custom step etotal temp vol press
thermo 1000
velocity all create $t 3627941 dist gaussian mom yes
velocity all scale $t
fix int all nvt temp $t $t 0.5 #between 0.2-2.0
run 5000
variable st equal step
variable tm equal step
variable Et equal etotal
variable Ep equal pe
variable Ek equal ke
variable tmp equal temp
variable pr equal press/10000
variable sxx equal pxx/10000
variable syy equal pyy/10000
variable szz equal pzz/10000
variable sxy equal pxy/10000
variable sxz equal pxz/10000
variable syz equal pyz/10000
fix statdump all print 100 "${st} ${tm} ${Et} ${Ep} ${Ek} ${tmp} ${pr} ${sxx} ${syy} ${szz} ${sxy} ${sxz} ${syz}" screen no file dump.stat
dump posdump all custom 100 dump.positions xs ys zs
dump_modify posdump sort id format line "%20.15e %20.15e %20.15e"
dump forcedump all custom 100 dump.forces fx fy fz
dump_modify forcedump sort id format line "%20.15e %20.15e %20.15e"
run 20000# molecular dynamics NVE
variable a index 3.4333879460894972
variable baseline equal 0.0
variable st equal step-5000
variable tm equal step-5000
variable Et equal etotal-v_baseline
variable Ep equal pe-v_baseline
variable Ek equal ke
variable tmp equal temp
variable pr equal press/10000
variable sxx equal pxx/10000
variable syy equal pyy/10000
variable szz equal pzz/10000
variable sxy equal pxy/10000
variable sxz equal pxz/10000
variable syz equal pyz/10000
units metal
dimension 3
boundary p p p # periodic boundary conditions (f ? aperiodic/ p - periodic)
atom_style atomic # Define what style of atoms to use in a simulation. http://lammps.sandia.gov/doc/atom_style.html
variable temperature equal 1113
variable temperature_x2 equal ${temperature}
lattice bcc ${a}
read_data data.pos
pair_style MLIP mlip.ini
pair_coeff * *
mass 1 238.029
neighbor 0.3 bin
neigh_modify every 20 delay 0 check no
#---------------------------------------
thermo 1
thermo_style custom step time temp etotal pe ke pxx pyy pzz pxy pxz pyz
velocity all create ${temperature_x2} 32000 rot yes mom yes
timestep 0.0005
fix 1 all nve
fix 2 all langevin ${temperature} ${temperature} 0.1 12345 zero yes
run 5000
unfix 2
fix statdump all print 100 "${st} ${tm} ${Et} ${Ep} ${Ek} ${tmp} ${pr} ${sxx} ${syy} ${szz} ${sxy} ${sxz} ${syz}" screen no file dump.stat
dump posdump all custom 100 dump.positions xs ys zs
dump forcedump all custom 100 dump.forces fx fy fz
dump_modify posdump sort id format line "%20.15e %20.15e %20.15e"
dump_modify forcedump sort id format line "%20.15e %20.15e %20.15e"
run 20000
注:需仔细检查infile.ssposcar 和 MD运行时的POSCAR是否一致,尤其是坐标。
运行命令
mpirun -np 16 lmp_mpi -in in.lammps得到 dump.stat dump.positions, dump.forces
运行 grep_dump.sh OR grep_dump2.sh
#!/bin/bash
# figure out how many atoms there are
na=`head -n 4 dump.forces | tail -n 1`
# remove the header from the stat file
grep -v '^#' dump.stat > infile.stat
# figure out how many timesteps there are
nt=`wc -l infile.stat | awk '{print $1}'`
# create the positions and force files
[ -f infile.forces ] && rm infile.forces
[ -f infile.positions ] && rm infile.positions
for t in `seq 1 ${nt}`
do
nl=$(( ${na}+9))
nll=$(( ${nl}*${t} ))
echo "t ${t} ${nl} ${nll}"
head -n ${nll} dump.forces | tail -n ${na} >> infile.forces
head -n ${nll} dump.positions | tail -n ${na} >> infile.positions
done
#grep_dump2.sh
#!/bin/bash
#number of atoms
Nat=$(head -n 4 dump.forces | tail -n 1)
#copy dump.stat to infile.stat
grep -v '^#' dump.stat > infile.stat
#number of timesteps
Nt=$(gawk 'END{print FNR}' infile.stat)
# create the positions and forces files
[ -f infile.forces ] && rm infile.forces
[ -f infile.positions ] && rm infile.positions
#prepare input files. Mind that first configuration in dump.forces and dump.positions should be excluded
((Nl=$Nt*(Nat+9)))
tail -n $Nl dump.forces | grep -A $Nat 'ITEM: ATOM' | grep -v 'ITEM: ATOM' | grep -v '^--$' >> infile.forces
tail -n $Nl dump.positions | grep -A $Nat 'ITEM: ATOM' | grep -v 'ITEM: ATOM' | grep -v '^--$' >> infile.positions得到infile.stat infile.positions infile.forces; 修改infile.meta的相关信息
自定义高对称点路径的脚本 infile.qpoints_dispersion
CUSTOM !
100 ! Number of points on each path
4 ! Number paths between special points
0.000 0.000 0.000 0.000 0.500 0.500 GM X
0.000 0.500 0.500 0.000 0.625 0.375 X U
0.375 0.750 0.375 0.000 0.000 0.000 K GM
0.000 0.000 0.000 0.000 0.500 0.000 GM LOR
FCC ! Bravais lattice type
100 ! Number of points on each path
4 ! Number paths between special points
GM X ! Starting and ending special point
X U !
K GM !
GM L !2 TDEP计算处理得到声子色散
简单的命令流程
extract_forceconstants -rc2 5
mv outfile.forceconstant infile.forceconstant
phonon_dispersion_relations
gnuplot outfile.dispersion_relations.gnuplot
Si的例子 VASP_MD
INCAR
Global Parameters
ISTART = 0 (Read existing wavefunction; if there)
# ISPIN = 2 (Spin polarised DFT)
# ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = Auto (Projection operators: automatic)
ENCUT = 400 (Cut-off energy for plane wave basis set, in eV)
PREC = Normal (Precision level)
LWAVE = .FALSE. (Write WAVECAR or not)
LCHARG = .FALSE. (Write CHGCAR or not)
#ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
# NWRITE = 2 (Medium-level output)
# KPAR = 2 (Divides k-grid into separate groups)
# NGX = 500 (FFT grid mesh density for nice charge/potential plots)
# NGY = 500 (FFT grid mesh density for nice charge/potential plots)
# NGZ = 500 (FFT grid mesh density for nice charge/potential plots)
GGA = PE
KPAR = 2
Electronic Relaxation
ISMEAR = -1
SIGMA = 0.02585201664
EDIFF = 1E-05
IALGO = 38
NELMIN = 4
Molecular Dynamics
IBRION = 0 (Activate MD)
NSW = 20000 (Max ionic steps)
#EDIFFG = -1E-03 (Ionic convergence; eV/A)
POTIM = 1 (Timestep in fs)
SMASS = 0 (MD Algorithm: -3-microcanonical ensemble; 0-canonical ensemble)
TEBEG = 300 (Start temperature K)
TEEND = 300 (Start temperature K)
MDALGO = 2 (Andersen Thermostat)
ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
ISIF = 2初始原胞
Primitive Cell
1.000000
0.00000000000000 2.73423319845426 2.73423319845426
2.73423319845426 0.00000000000000 2.73423319845426
2.73423319845426 2.73423319845426 0.00000000000000
Si
2
DIRECT
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000超胞为3x3x3
运行VASP进行分子动力学计算
运行process_outcar_5.3.py 提取产生TDEP输入文件
mpirun -np 16 vasp_std
process_outcar_5.3.py OUTCAR --skip n # n为第几步开始提取注:需仔细检查infile.ssposcar 和 MD运行时的POSCAR是否一致,尤其是坐标。
TDEP步骤
extract_forceconstants -rc2 5 -rc3 3
mv outfile.forceconstant infile.forceconstant
#phonon dispersion
phonon_dispersion_relations
#phonon lineshape
mv outfile.infile.forceconstant_thirdorder infile.forceconstant_thirdorder
lineshape --path -qg 12 12 12 -ne 600 --temperature 300
lineshape的画图脚本
import matplotlib.pyplot as plt
import numpy as np
import h5py as h5
from matplotlib.colors import LogNorm
# open the sqe file
f = h5.File('outfile.sqe.hdf5','r')
# get axes and intensity
x = np.array(f.get('q_values'))
y = np.array(f.get('energy_values'))
gz = np.array(f.get('intensity'))
# add a little bit so that the logscale does not go nuts
gz=gz+1E-2
# for plotting, turn the axes into 2d arrays
gx, gy = np.meshgrid(x,y)
# x-ticks
xt = np.array(f.get('q_ticks'))
# labels for the x-ticks
xl = f.attrs.get('q_tick_labels').split()
# label for y-axis
plt.pcolormesh(gx, gy, gz, norm=LogNorm(vmin=gz.min(), vmax=gz.max()), cmap='afmhot')
# set the limits of the plot to the limits of the data
plt.axis([x.min(), x.max(), y.min(), y.max()])
plt.xticks(xt,xl)
plt.tight_layout()
plt.show()