电子结构的计算流程
高对称点生成和后期处理使用vaspkit-1.1。
注:该方法只针对原胞。
PBE能带计算流程
必要的文件
INCAR-scf
INCAR-band
KPOINTS
POTCAR
POSCAR #优化后的原胞
run_banddos.shrun_banddos.sh的脚本设置如下:
#!/bin/bash
#优化后的初始POSCAR
cat > POSCAR <<!
Primitive Cell
1.000000
0.00000000000000 2.73423319845426 2.73423319845426
2.73423319845426 0.00000000000000 2.73423319845426
2.73423319845426 2.73423319845426 0.00000000000000
Si
2
DIRECT
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000
!
cat > KPOINTS <<!
A
0
M
12 12 12
0 0 0
!
#基于seek-path自动生成高对称点
vaspkit -task 303
#得到KPATH.in 和 HIGH_SYMMETRY_POINTS 可以自行修改
cat > INCAR-scf <<!
ISTART=0
ICHARG=2
PREC=Accurate
GGA = PE
ADDGRID =.TRUE.
# Electronic relaxation
ENCUT=500
EDIFF=1E-8
EDIFFG=-0.001
ISMEAR=0
SIGMA = 0.05
POTIM=0.20
# Other Tags
#PSTRESS=
# Write flags
LWAVE=.TRUE.
LCHARG=.TRUE.
!
cat > INCAR-band <<!
Global Parameters
ISTART = 1 (Read existing wavefunction; if there)
ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = F (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing; metals:1)
SIGMA = 0.05 (Smearing value in eV; metals:0.2)
# NELM = 60 (Max electronic SCF steps)
# NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
LORBIT=11 (plot projection band need to set)
NEDOS = 2000
Ionic Relaxation
# NELMIN = 6 (Min electronic SCF steps)
NSW = 0 (Max electronic SCF steps)
IBRION = -1 (Algorithm: 0-MD; 1-Quasi-New; 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -0.001 (Ionic convergence; eV/AA)
#ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
!
#自洽计算
cp INCAR-scf INCAR
echo "scf"; time mpirun -np 16 vasp_std
rm INCAR
#更换KPOINTS文件
cp KPATH.in KPOINTS
cp INCAR-band INCAR
echo "band"; time mpirun -np 16 vasp_std
#处理能带数据
vaspkit -task n #根据下面说明自行选择
### 211) Band-Structure
### 212) Projected Band-Structure for Selected Atom
### 213) Projected Band-Structure for Each Element
### 214) The Sum of Projected Band-Structure for Selected Atoms
### 215) Projected Band-Structure by Element Weight
###运行后得到包含能带数据的dat文件
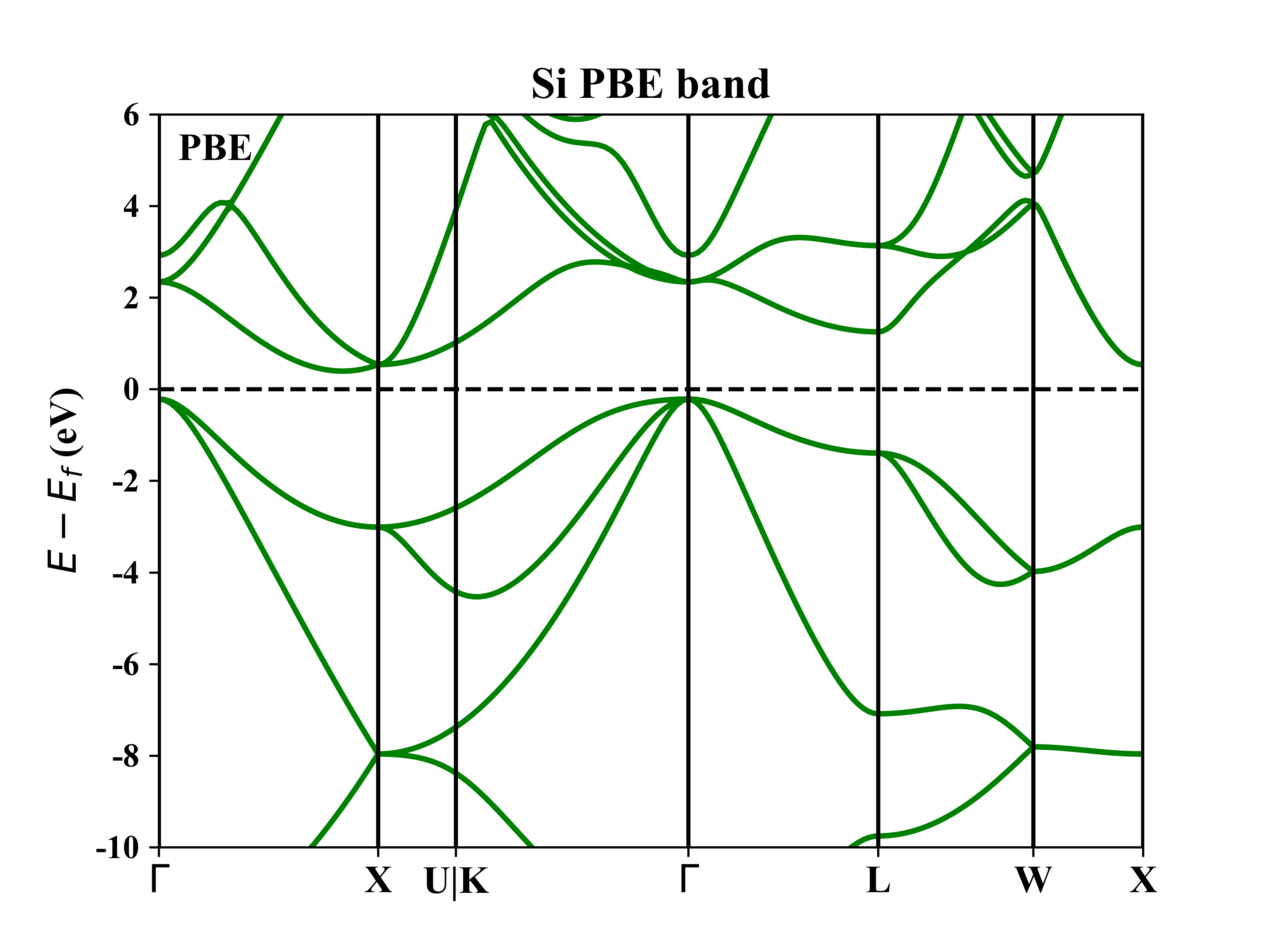
Si的能带图:
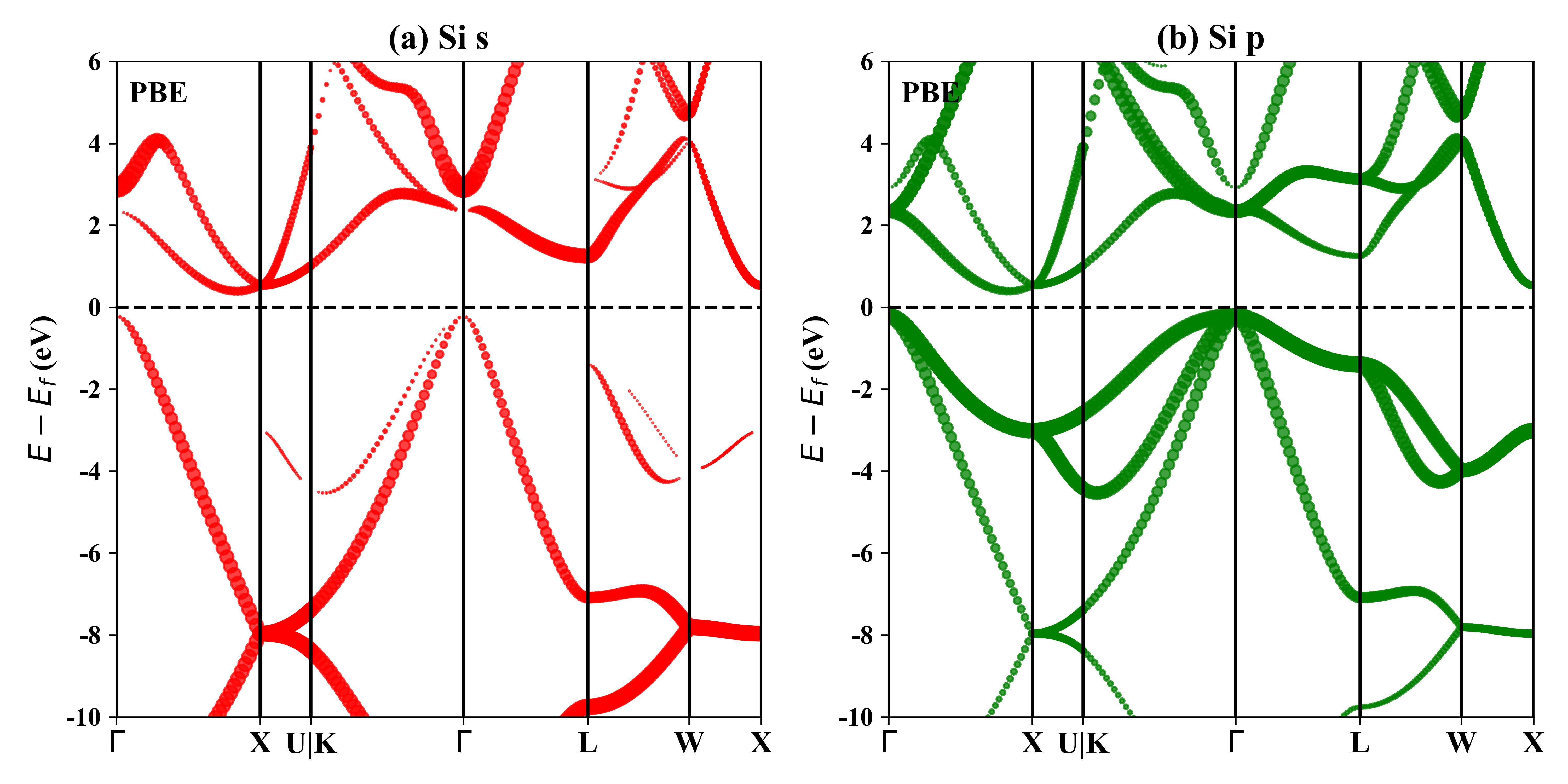
Si的轨道投影能带:
杂化轨道能带计算流程
必要的文件
INCAR-scf
INCAR-hse
KPOINTS
POTCAR
POSCAR #优化后的原胞
run_hseband.shrun_hseband.sh脚本设置如下:
#!/bin/bash
#优化后的初始POSCAR
cat > POSCAR <<!
Primitive Cell
1.000000
0.00000000000000 2.73423319845426 2.73423319845426
2.73423319845426 0.00000000000000 2.73423319845426
2.73423319845426 2.73423319845426 0.00000000000000
Si
2
DIRECT
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000
!
#基于seek-path自动生成高对称点
vaspkit -task 303
#得到KPATH.in 和 HIGH_SYMMETRY_POINTS 可以自行修改
#由于HSE计算量很大,对于点的数量一定要适当
vaspkit -task 251
#生成包含SCF的k-mesh和权重为0的用于HSE-band的k-path的KPOINTS文件
cat > INCAR-scf <<!
ISTART=0
ICHARG=2
PREC=Accurate
GGA = PE
ADDGRID =.TRUE.
# Electronic relaxation
ENCUT=500
EDIFF=1E-8
EDIFFG=-0.001
ISMEAR=0
SIGMA = 0.05
POTIM=0.20
# Other Tags
#PSTRESS=
# Write flags
LWAVE=.TRUE.
LCHARG=.TRUE.
!
cat > INCAR-hse <<!
Global Parameters
ISTART = 1 (Read existing wavefunction; if there)
ICHARG = 1 (Non-self-consistent: GGA/LDA band structures)
LREAL = F (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing; metals:1)
SIGMA = 0.05 (Smearing value in eV; metals:0.2)
# NELM = 60 (Max electronic SCF steps)
# NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
LORBIT=11
NEDOS = 5000
Ionic Relaxation
# NELMIN = 6 (Min electronic SCF steps)
NSW = 0 (Max electronic SCF steps)
IBRION = -1 (Algorithm: 0-MD; 1-Quasi-New; 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -0.001 (Ionic convergence; eV/AA)
ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
#HSE06 Calculation
LHFCALC= .TRUE. (Activate HF)
AEXX = 0.25 (25% HF exact exchange, adjusted this value to reproduce experimental band gap)
HFSCREEN= 0.2 (Switch to screened exchange; e.g. HSE06)
ALGO = Damped (Electronic Minimisation Algorithm; ALGO=58)
TIME = 0.4 (Timestep for IALGO5X)
PRECFOCK= Normal (HF FFT grid)
# NKRED = 2 (Reduce k-grid-even only, see also NKREDX, NKREDY and NKREDZ)
# HFLMAX = 4 (HF cut-off: 4d, 6f)
# LDIAG = .TRUE. (Diagnolise Eigenvalues)
!
#SCF计算,可省略,主要得到WAVECAR波函数,可加速HSE计算速度
cp INCAR-scf INCAR
echo "scf"; time mpirun -np 16 vasp_std
rm INCAR
#HSE计算
cp INCAR-hse INCAR
echo "hse"; time mpirun -np 16 vasp_std
vaspkit -task n #根据下述说明自行选择
## 250) Generate KPOINTS Including Irreducible Kmesh and Band Edges
## 251) Generate KPOINTS File for Hybrid Band-Structure Calculation
## 252) Get Band-Structure for Hybrid-DFT Calculation
## 253) Get Projected Band-Structure for Selected Atoms
## 254) Get Projected Band-Structure for Each ELement
## 255) Get the Sum of Projected Band-Structure for Selected Atoms
## 256) Projected Band-Structure by Element Weight
PBE(HSE)+SOC计算能带
参照 PBE(HSE)计算能带。
在INCAR中添加如下参数:
Spin-Orbit Coupling Calculation
ISPIN = 2
NELMIN = 6 (Min electronic SCF steps)
LSORBIT = .TRUE. (Activate SOC)
GGA_COMPAT = .FALSE. (Apply spherical cutoff on gradient field)
# VOSKOWN = 1 (Enhances the magnetic moments and the magnetic energies)
LMAXMIX = 2 (For d elements increase LMAXMIX to 4, f: LMAXMIX = 6)
SAXIS = 0 0 1 (Direction of the magnetic field)
# MAGMOM = 0 0 3 (Set this parameters manually, Local magnetic moment parallel to SAXIS, 3*NIONS*1.0 for non-collinear magnetic systems)
NBANDS = 32 (Set this parameters manually, 2 * number of bands of collinear-run)注:NBAND最好为PBE(HSE)中能带数的整数倍。HSE+SOC计算流程未测试,不知道会不会成功。下面是我当初计算2D-Tl2O的HSE+SOC能带的流程,主要给出INCAR的设置。
第一步:SCF+SOC计算
Global Parameters
ISTART = 0 (Read existing wavefunction; if there)
ICHARG = 2 (Non-self-consistent: GGA/LDA band structures)
LREAL = F (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing; metals:1)
SIGMA = 0.05 (Smearing value in eV; metals:0.2)
# NELM = 60 (Max electronic SCF steps)
# NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-06 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
Ionic Relaxation
# NELMIN = 6 (Min electronic SCF steps)
NSW = 0 (Max electronic SCF steps)
IBRION = -1 (Algorithm: 0-MD; 1-Quasi-New; 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -0.01 (Ionic convergence; eV/AA)
ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
Spin-Orbit Coupling Calculation
ISPIN = 2
NELMIN = 6 (Min electronic SCF steps)
LSORBIT = .TRUE. (Activate SOC)
GGA_COMPAT = .FALSE. (Apply spherical cutoff on gradient field)
# VOSKOWN = 1 (Enhances the magnetic moments and the magnetic energies)
LMAXMIX = 2 (For d elements increase LMAXMIX to 4, f: LMAXMIX = 6)
SAXIS = 0 0 1 (Direction of the magnetic field)
# MAGMOM = 0 0 3 (Set this parameters manually, Local magnetic moment parallel to SAXIS, 3*NIONS*1.0 for non-collinear magnetic systems)
# NBANDS = 32 (Set this parameters manually, 2 * number of bands of collinear-run)
得到IBZKPT的K点
能带计算K点的生成参照HSE计算
第二步:打开杂化HSE
Global Parameters
ISTART = 1 (Read existing wavefunction; if there)
ICHARG = 2 (Non-self-consistent: GGA/LDA band structures)
LREAL = F (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing; metals:1)
SIGMA = 0.15 (Smearing value in eV; metals:0.2)
# NELM = 60 (Max electronic SCF steps)
# NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-06 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
Ionic Relaxation
# NELMIN = 6 (Min electronic SCF steps)
NSW = 0 (Max electronic SCF steps)
IBRION = -1 (Algorithm: 0-MD; 1-Quasi-New; 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -0.01 (Ionic convergence; eV/AA)
ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
Spin-Orbit Coupling Calculation
ISPIN = 2
NELMIN = 6 (Min electronic SCF steps)
LSORBIT = .TRUE. (Activate SOC)
GGA_COMPAT = .FALSE. (Apply spherical cutoff on gradient field)
# VOSKOWN = 1 (Enhances the magnetic moments and the magnetic energies)
LMAXMIX = 2 (For d elements increase LMAXMIX to 4, f: LMAXMIX = 6)
SAXIS = 0 0 1 (Direction of the magnetic field)
# MAGMOM = 0 0 3 (Set this parameters manually, Local magnetic moment parallel to SAXIS, 3*NIONS*1.0 for non-collinear magnetic systems)
NBANDS = 32 (Set this parameters manually, 2 * number of bands of collinear-run)
HSE06 Calculation
LHFCALC= .TRUE. (Activate HF)
AEXX = 0.25 (25% HF exact exchange, adjusted this value to reproduce experimental band gap)
HFSCREEN= 0.2 (Switch to screened exchange; e.g. HSE06)
ALGO = Damped (Electronic Minimisation Algorithm; ALGO=58)
TIME = 0.4 (Timestep for IALGO5X)
PRECFOCK= Normal (HF FFT grid)
# NKRED = 2 (Reduce k-grid-even only, see also NKREDX, NKREDY and NKREDZ)
# HFLMAX = 4 (HF cut-off: 4d, 6f)
# LDIAG = .TRUE. (Diagnolise Eigenvalues) 注:选择读取上一步的WAVECAR可以加速这一步的计算,如果报错,可以不读取WAVECAR直接计算。
态密度计算
注:与能带计算差不多,只是不需要修改K点。也可以直接用能带计算中得到的态密度。
必要的文件:
INCAR-scf
INCAR-dos
KPOINTS
POTCAR
run_dos.sh#!/bin/bash
cat > POSCAR <<!
Primitive Cell
1.000000
0.00000000000000 2.73423319845426 2.73423319845426
2.73423319845426 0.00000000000000 2.73423319845426
2.73423319845426 2.73423319845426 0.00000000000000
Si
2
DIRECT
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000
!
cat > KPOINTS <<!
A
0
M
21 21 21
0 0 0
!
cat > INCAR-scf <<!
ISTART=0
ICHARG=2
PREC=Accurate
GGA = PE
ADDGRID =.TRUE.
# Electronic relaxation
ENCUT=500
EDIFF=1E-8
EDIFFG=-0.001
ISMEAR=0
SIGMA = 0.05
POTIM=0.20
# Other Tags
#PSTRESS=
# Write flags
LWAVE=.TRUE.
LCHARG=.TRUE.
!
cat > INCAR-dos <<!
Global Parameters
ISTART = 1 (Read existing wavefunction; if there)
ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = F (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells; be careful)
# LPLANE = .TRUE. (Real space distribution; supercells)
# NPAR = 4 (Max is no. nodes; don't set for hybrids)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing; metals:1)
SIGMA = 0.05 (Smearing value in eV; metals:0.2)
# NELM = 60 (Max electronic SCF steps)
# NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
LORBIT=11 (plot projection band need to set)
NEDOS = 2000
Ionic Relaxation
# NELMIN = 6 (Min electronic SCF steps)
NSW = 0 (Max electronic SCF steps)
IBRION = -1 (Algorithm: 0-MD; 1-Quasi-New; 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -0.001 (Ionic convergence; eV/AA)
#ISYM = 0 (Symmetry: 0=none; 2=GGA; 3=hybrids)
!
#自洽计算
cp INCAR-scf INCAR
echo "scf"; time mpirun -np 16 vasp_std
rm INCAR
#更换KPOINTS文件
cp INCAR-dos INCAR
echo "dos"; time mpirun -np 16 vasp_std
vaspkit -task n #根据需要选择下面的参数,参照手册说明,有些需要给出元素,个数
### 111) Total Density-of-States
### 112) Projected Density-of-States for Selected Atoms
### 113) Projected Density-of-States for Each Element
### 114) The Sum of Projected Density-of-States for Selected Atoms
### 115) The Sum of Projected DOS for Selected Atoms and Orbitals
### 116) Local Density-of-States for Each Element
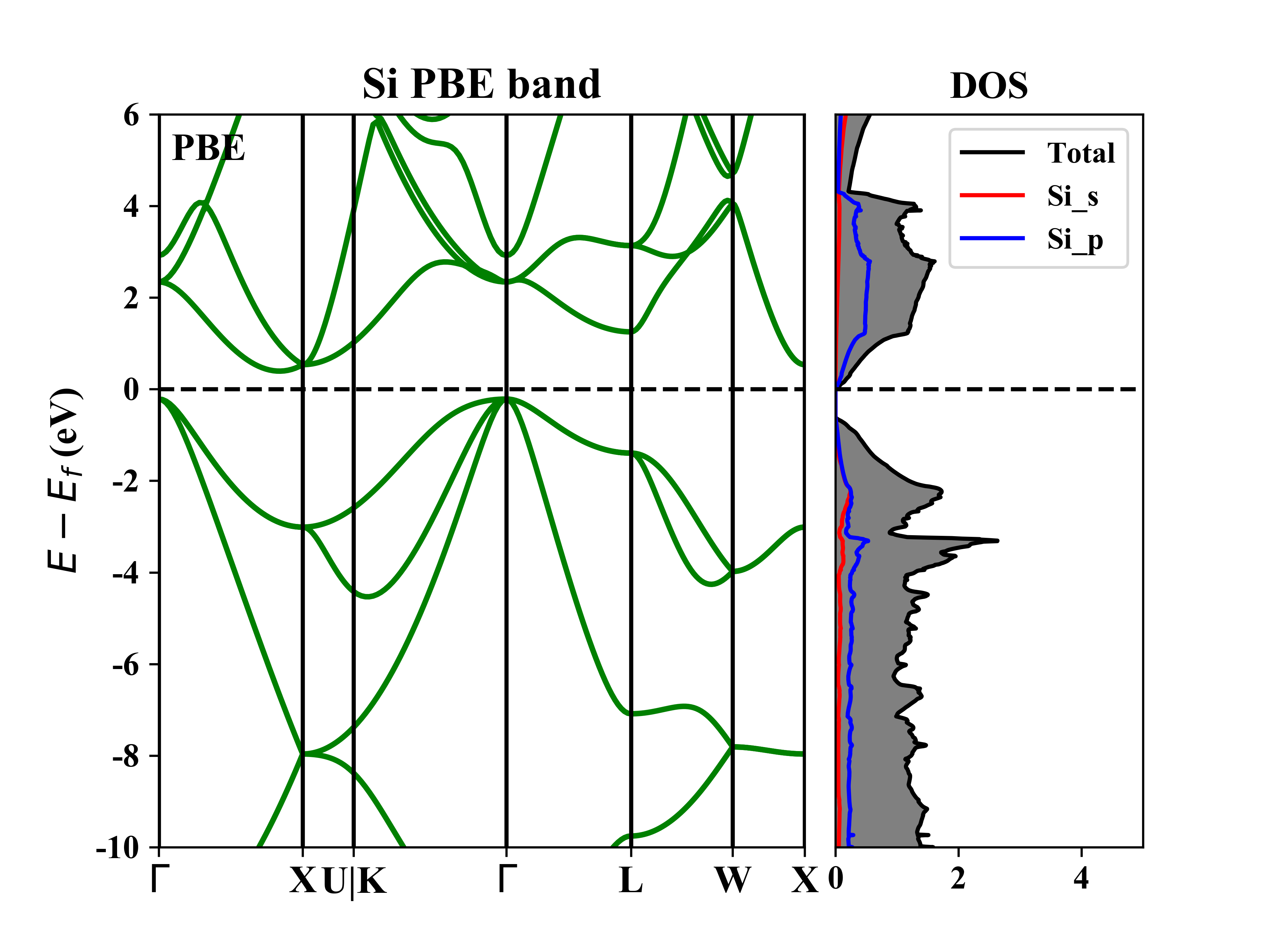
### 117) Total Density-of-States with Adjustable Smearing WidthSi的band-dos图如下:
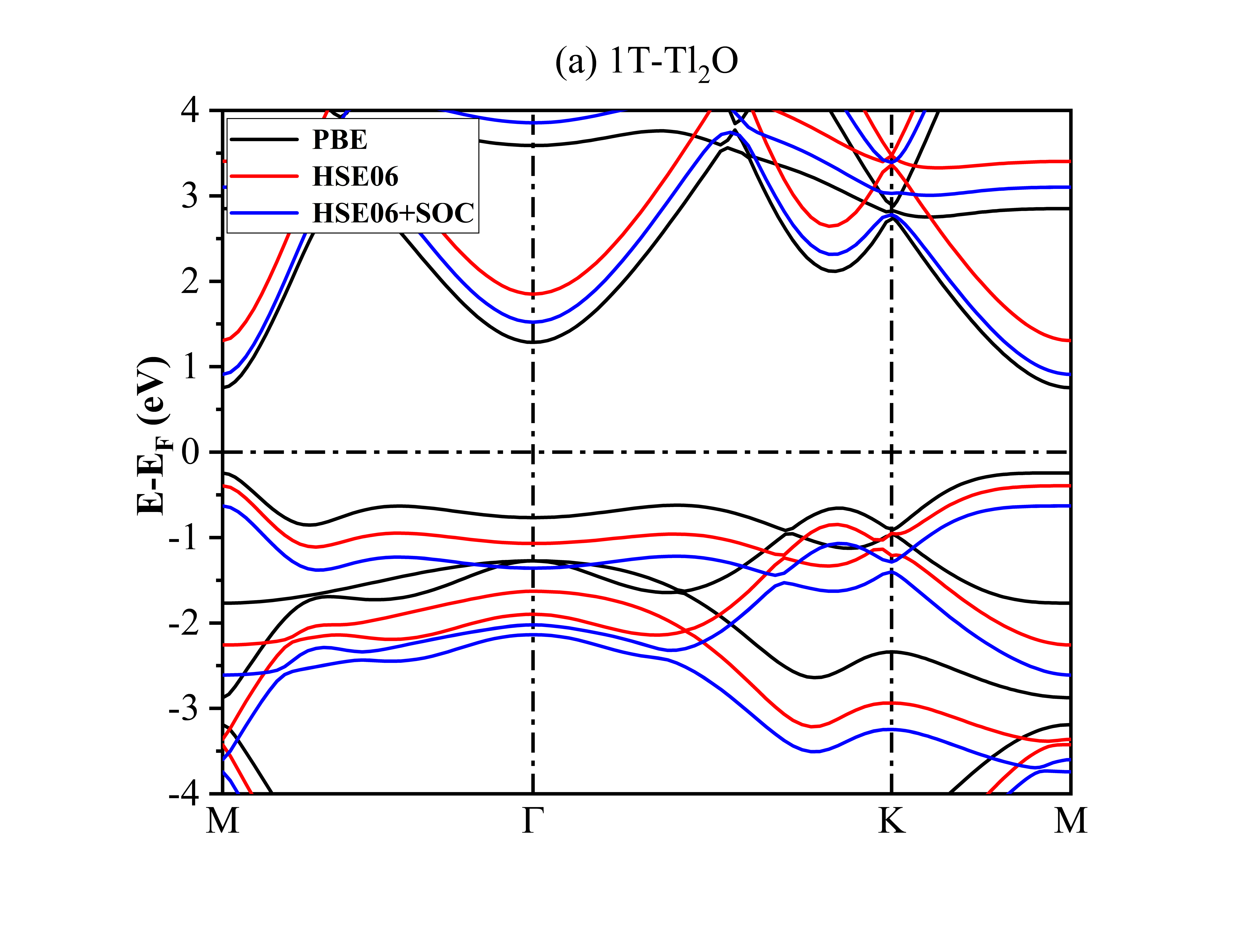
二维材料1T-Tl2O的能带示例
PBE
1T-Tl2O的能带态密度:
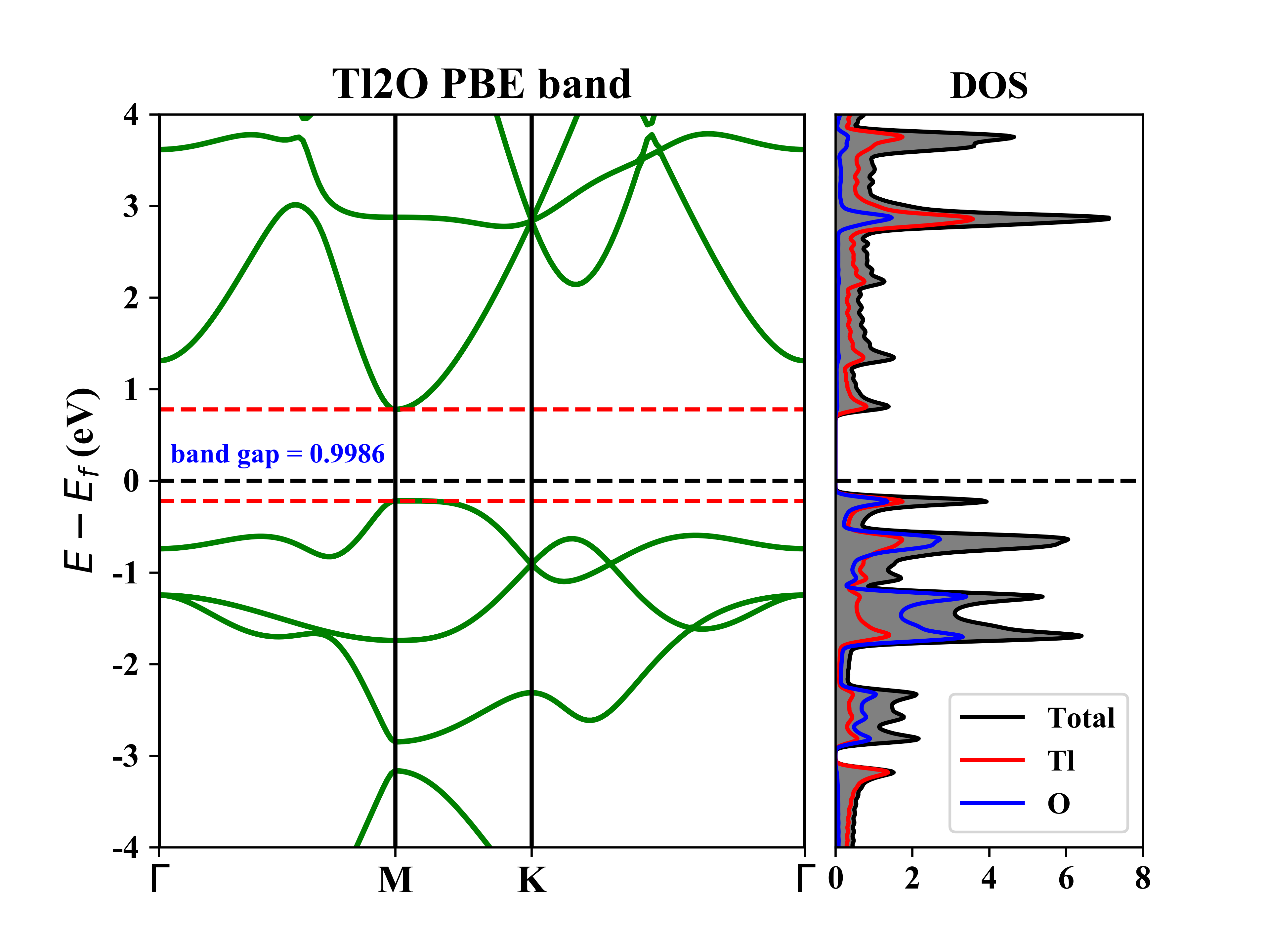
1T-Tl2O的投影能带:
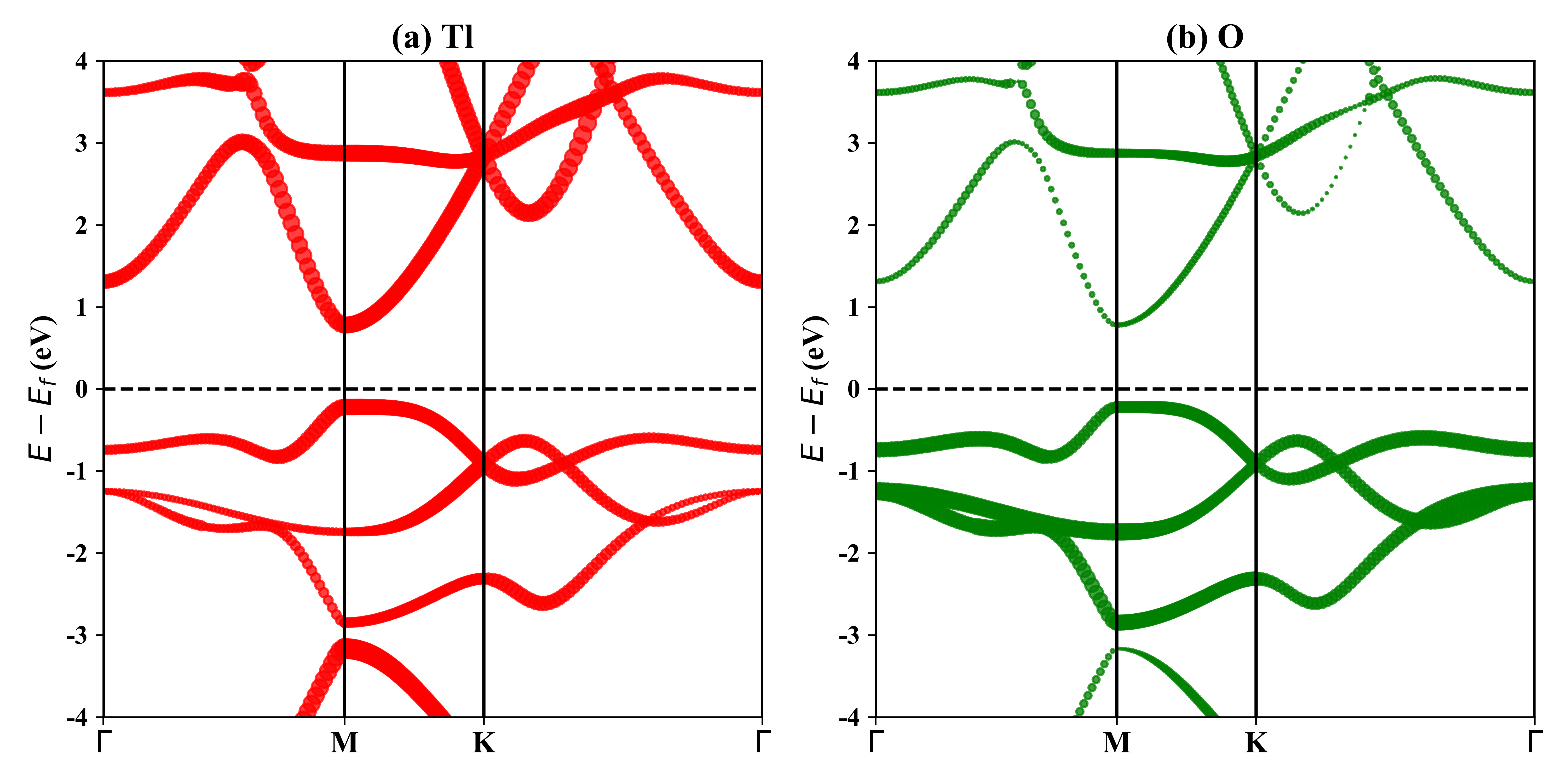
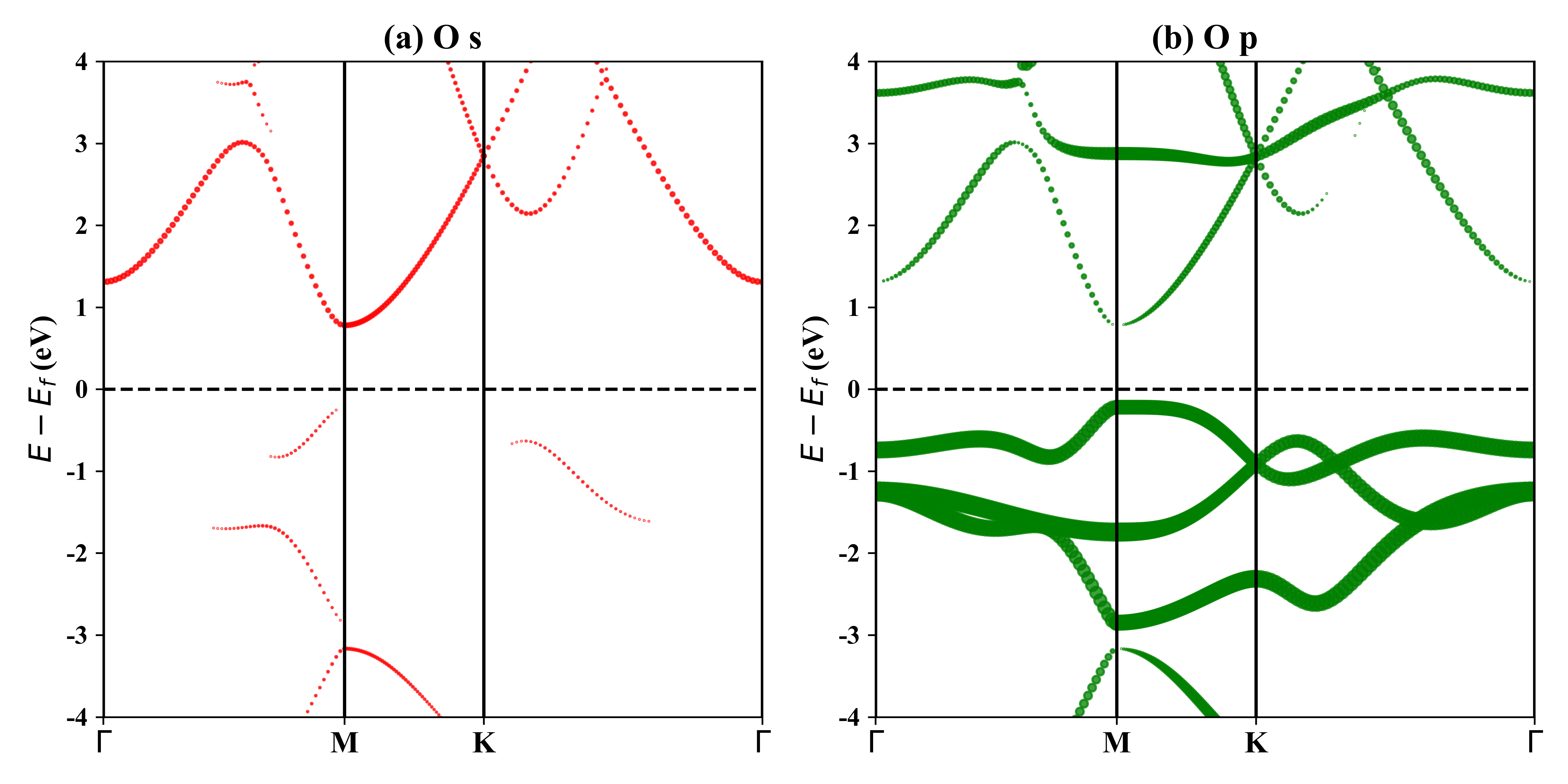
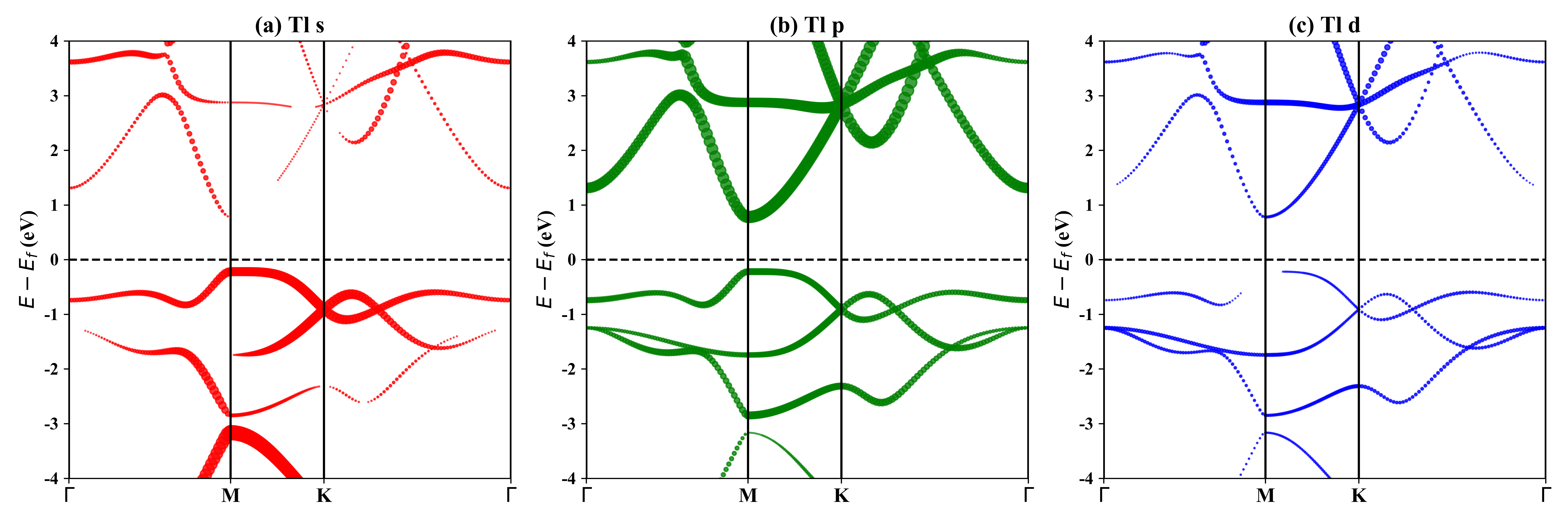
HSE
1T-Tl2O的能带态密度:
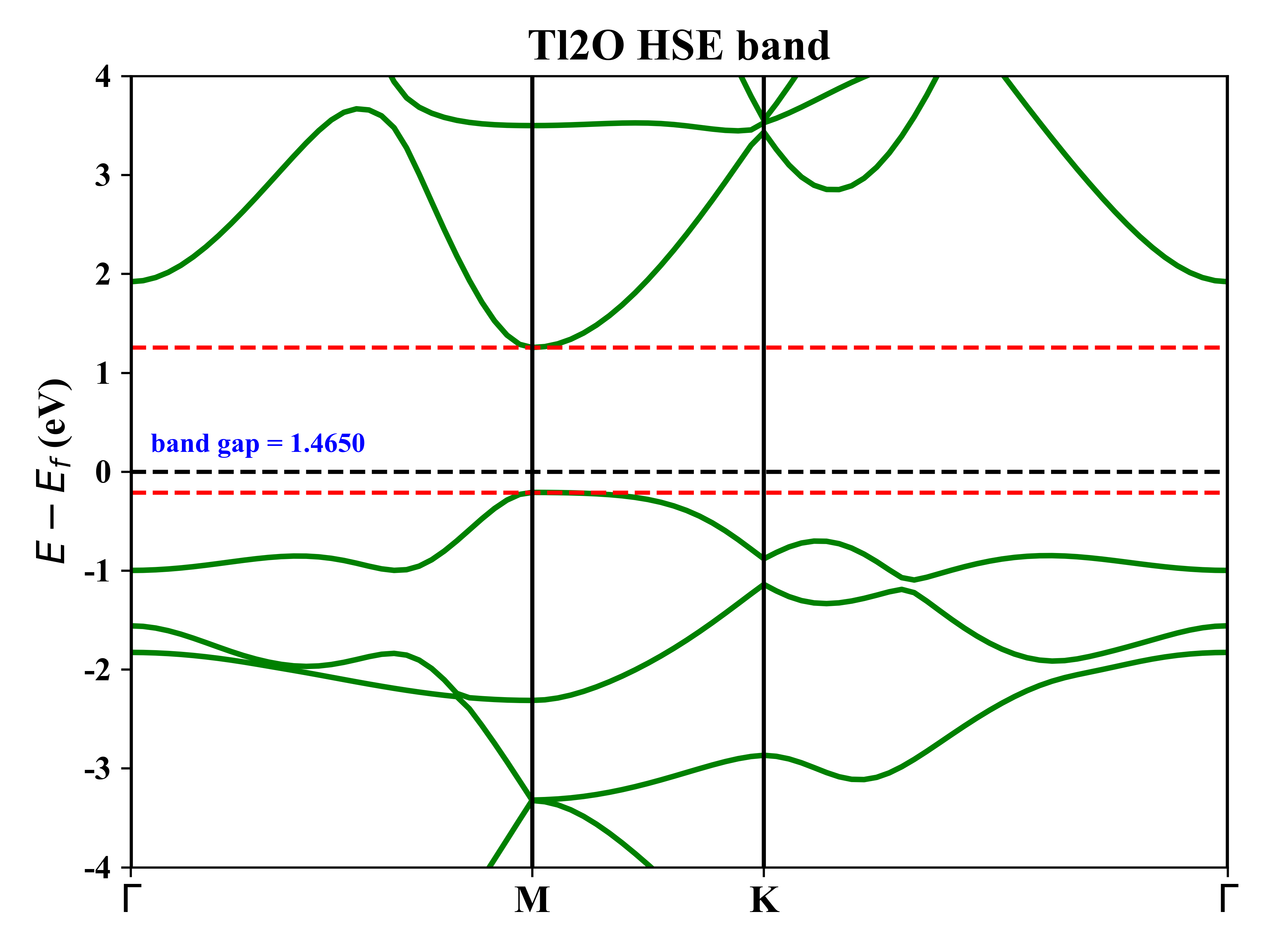
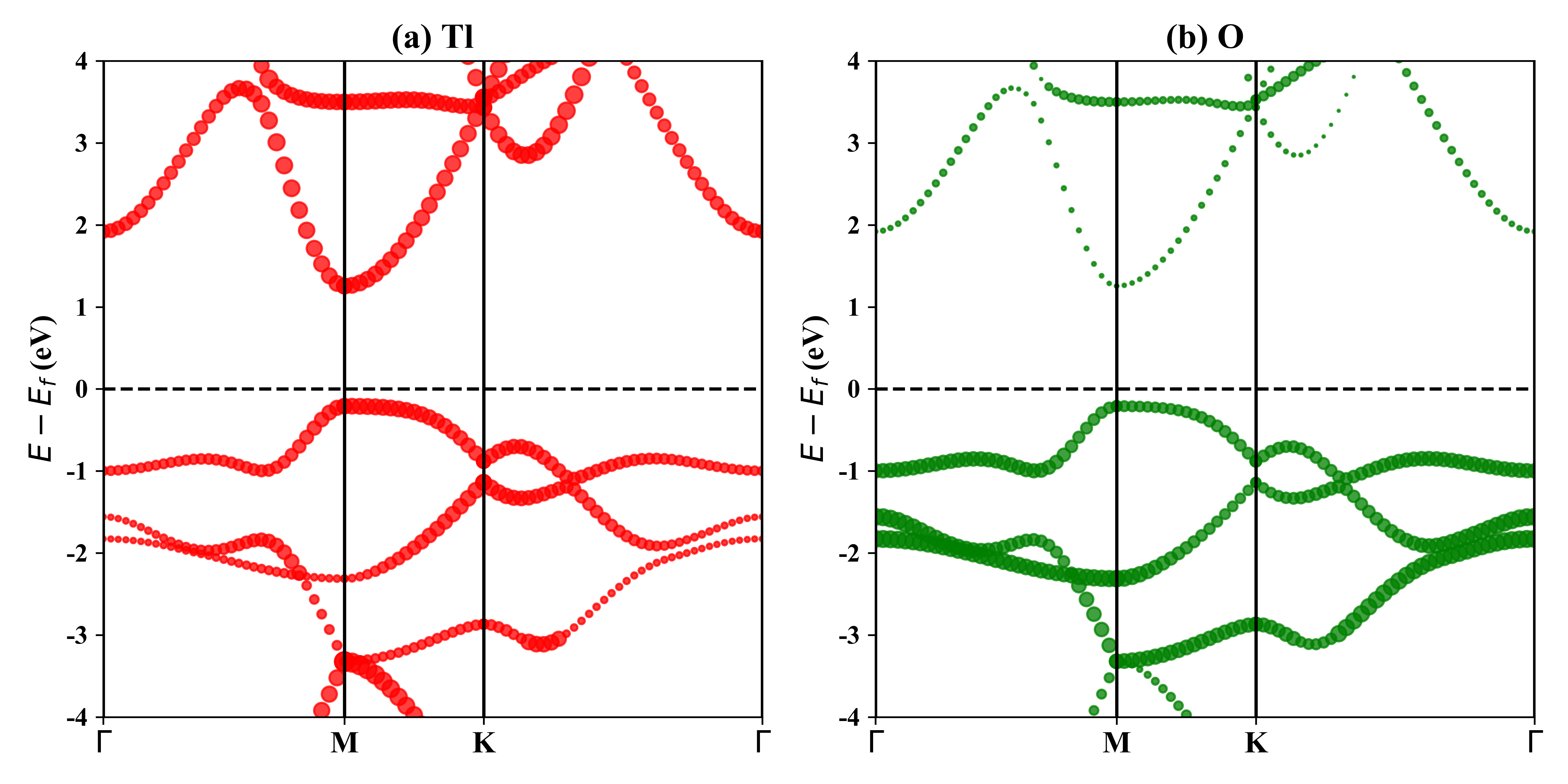
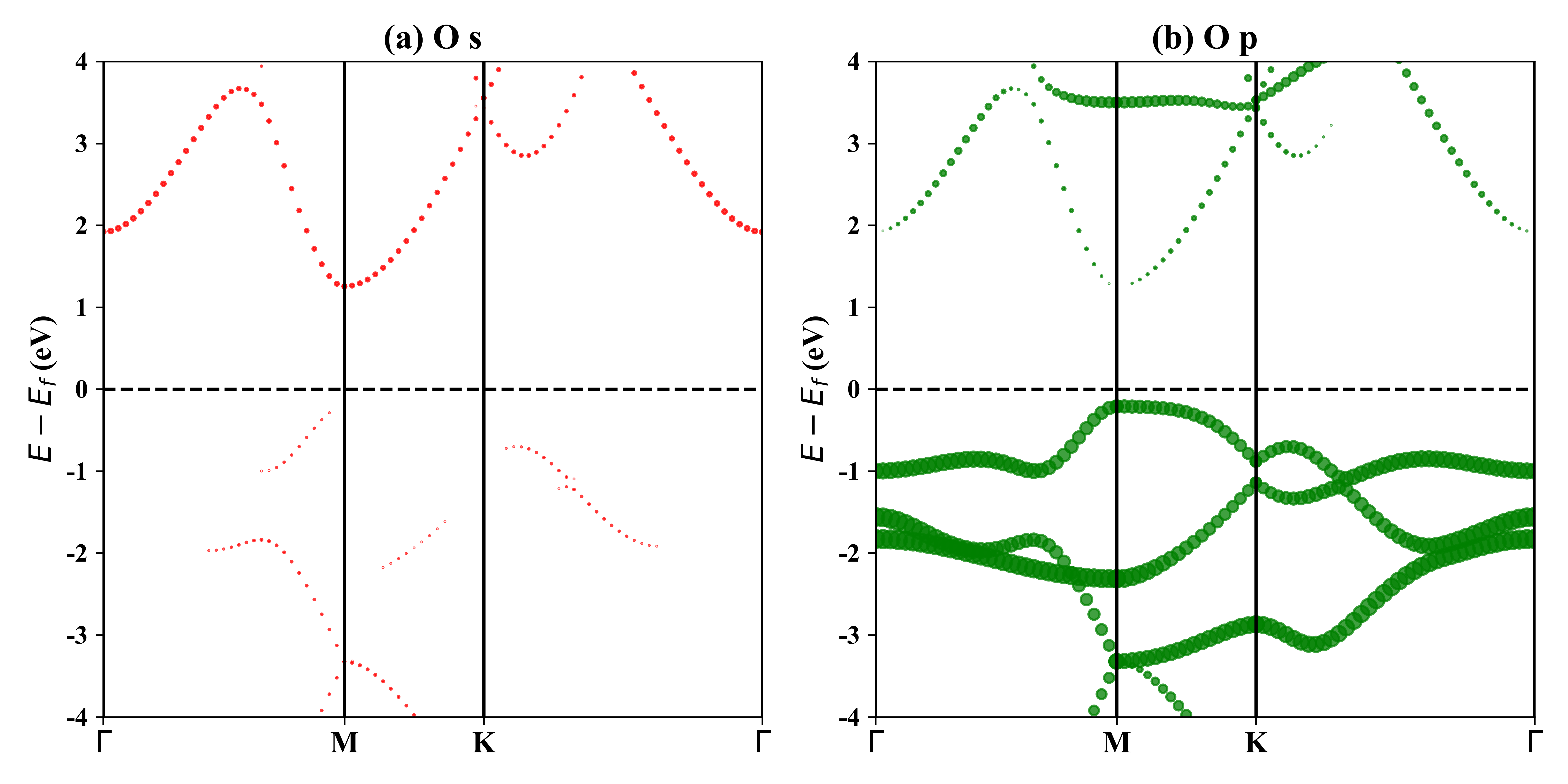
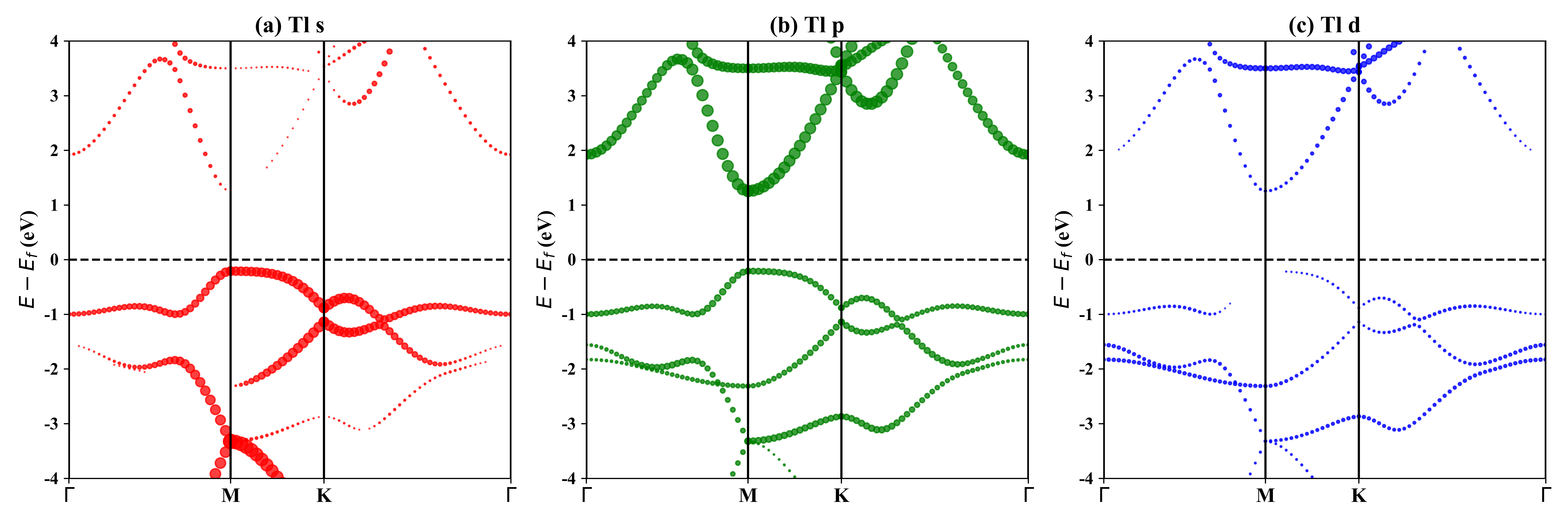
1T-Tl2O的投影能带:
HSE+SOC