KPOINTS 或 KSPACING 测试
必要输入文件:
k_test.sh
POTCAR
INCAR和POSCAR可在k_test.sh中设置,也可独立给出脚本1(k_test.sh): KPOINTS测试
#!/bin/bash
#生成vasp静态计算的INCAR
cat > INCAR_static <<!
Global Parameters
ISTART = 0 (Read existing wavefunction; if there)
ICHARG = 2 (Non-self-consistent: GGA/LDA band structures)
LREAL = .FALSE. (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .FALSE. (Write WAVECAR or not)
LCHARG = .FALSE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
Electronic Relaxation
ISMEAR = -5 (Gaussian smearing; metals:1)
#SIGMA = 0.05 (Smearing value in eV; metals:0.2)
NELM = 60 (Max electronic SCF steps)
NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-06 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
Ionic Relaxation
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V,4-Shape/Ions)
EDIFFG = -0.001 (Ionic convergence; eV/AA)
KSPACING = 0.10
!
cp INCAR_static INCAR
#产生计算所需POSCAR
cat > POSCAR<<!
Si8
1.0000000000
5.4687280000 0.0000000000 0.0000000000
0.0000000000 5.4687280000 0.0000000000
0.0000000000 0.0000000000 5.4687280000
Si
8
Direct
0.0000000000 0.0000000000 0.0000000000
0.2500000000 0.7500000000 0.7500000000
0.5000000000 0.0000000000 0.5000000000
0.0000000000 0.5000000000 0.5000000000
0.5000000000 0.5000000000 0.0000000000
0.7500000000 0.2500000000 0.7500000000
0.7500000000 0.7500000000 0.2500000000
0.2500000000 0.2500000000 0.2500000000
!
for i in $(seq 6 3 15) #K点从6-24,步数为3
do
#生成KPOINTS文件,K点类型:Monkhorst-Pack (M) 或者 Gamma (G)
cat > KPOINTS <<!
Automatic mesh
0
Monkhorst-Pack
$i $i $i
0 0 0
!
echo "KPOINTS = $i" ; time mpirun -n 16 vasp_std #vasp并行运行命令,根据系统自行修改
rm KPOINTS
#提取计算得到的能量
E=$(grep "TOTEN" OUTCAR | tail -1 | awk '{printf "%12.9f \n", $5 }')
echo $i $E >>kpoints_energy.out
done
提交脚本任务运行,成功结束后可以得到输出文件kpoints_enengy.out
可以使用OriginPro等画图工具画出kpoints-能量关系
Si的KPOINTS测试
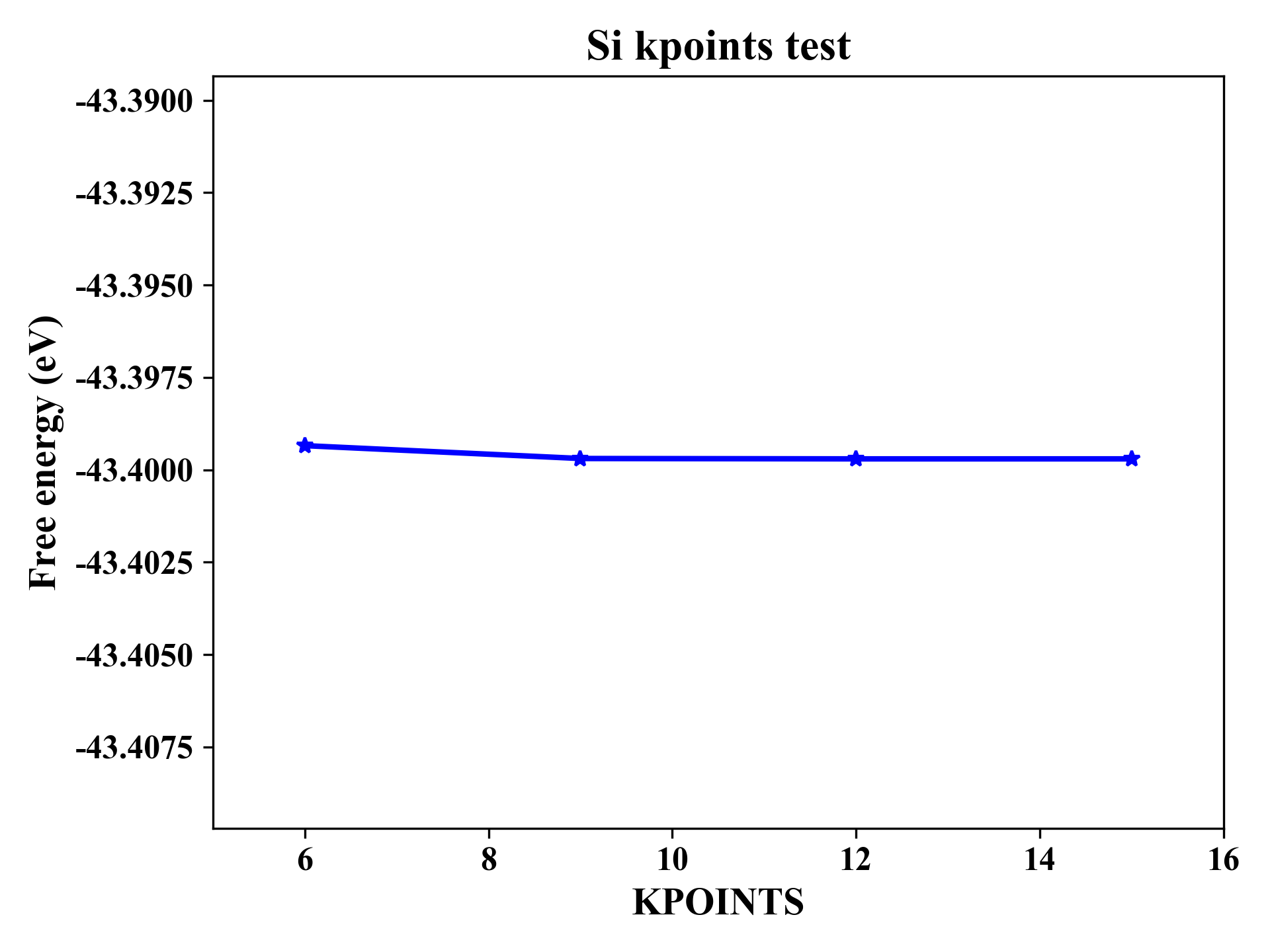
根据收敛性,大概选择KPOINTS为$9\times9\times9$
脚本2(k_test.sh): KSPACING测试
#!/bin/bash
#产生计算所需POSCAR
cat > POSCAR<<!
Si8
1.0000000000
5.4687280000 0.0000000000 0.0000000000
0.0000000000 5.4687280000 0.0000000000
0.0000000000 0.0000000000 5.4687280000
Si
8
Direct
0.0000000000 0.0000000000 0.0000000000
0.2500000000 0.7500000000 0.7500000000
0.5000000000 0.0000000000 0.5000000000
0.0000000000 0.5000000000 0.5000000000
0.5000000000 0.5000000000 0.0000000000
0.7500000000 0.2500000000 0.7500000000
0.7500000000 0.7500000000 0.2500000000
0.2500000000 0.2500000000 0.2500000000
!
for i in $(seq 0.5 -0.05 0.1) #K点密度从0.5-0.1,步数为0.05
do
#生成vasp静态计算的INCAR
cat > INCAR_static <<!
Global Parameters
ISTART = 0 (Read existing wavefunction; if there)
ICHARG = 2 (Non-self-consistent: GGA/LDA band structures)
LREAL = .FALSE. (Projection operators: automatic)
ENCUT = 500 (Cut-off energy for plane wave basis set, in eV)
PREC = Accurate (Precision level)
LWAVE = .FALSE. (Write WAVECAR or not)
LCHARG = .FALSE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid; helps GGA convergence)
Electronic Relaxation
ISMEAR = -5 (Gaussian smearing; metals:1)
#SIGMA = 0.05 (Smearing value in eV; metals:0.2)
NELM = 60 (Max electronic SCF steps)
NELMIN = 4 (Min electronic SCF steps)
EDIFF = 1E-06 (SCF energy convergence; in eV)
GGA = PE (PBEsol exchange-correlation)
Ionic Relaxation
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V,4-Shape/Ions)
EDIFFG = -0.001 (Ionic convergence; eV/AA)
KSPACING = $i
KGAMMA = .FALSE. #(true: GAMMA; false: Monkhorst-Pack )
!
cp INCAR_static INCAR
echo "KPOINTS = $i" ; time mpirun -n 16 vasp_std #vasp并行运行命令,根据系统自行修改
rm INCAR_static
rm INCAR
#提取计算得到的能量
E=$(grep "TOTEN" OUTCAR | tail -1 | awk '{printf "%12.9f \n", $5 }')
echo $i $E >>kspace_energy.out
done
注:Auto mesh模式下。 $KPONITSnumber = max(1,|\vec{b}|/(2\pi\times{KSPACING}))$
另外,后续介绍中,主要使用的是KPOINTS文件,KSPACING可以根据自行需要选择使用。
提交脚本任务运行,成功结束后可以得到输出文件kspace_enengy.out
可以使用OriginPro等画图工具画出k点密度-能量关系。
Si的KSPACE测试
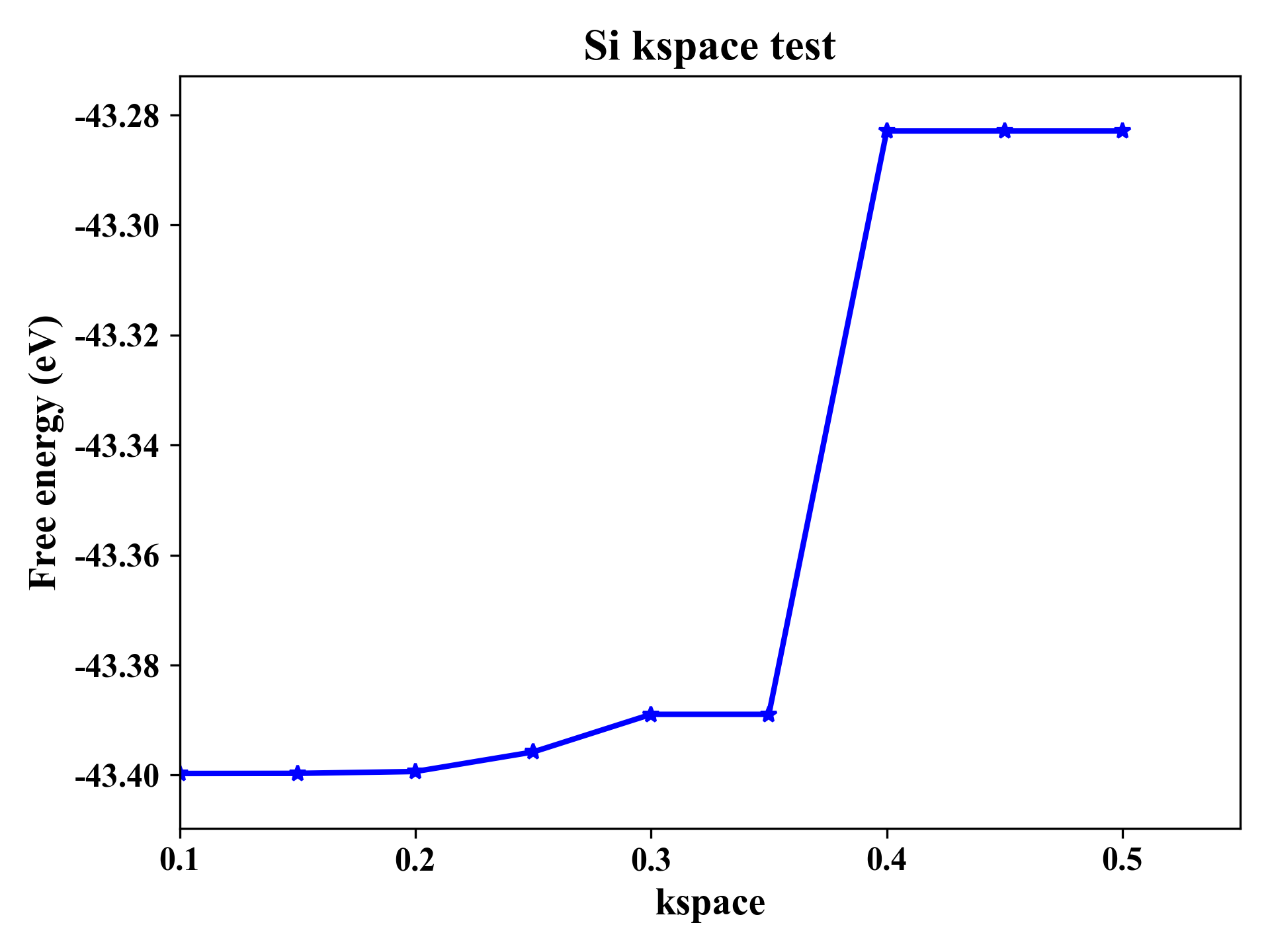
根据能量收敛判断,选择K点密度为0.15较为合适。

